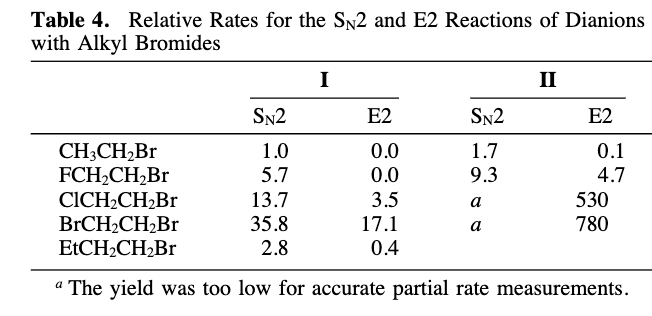
) Consider Table 4 and suggest why FCH 2 CH 2 Br (5.7 vs. 1) has a higher S N 2 reaction rate than EtBr. 4b) Consider Table 4 and suggest why BrCH 2 CH 2 Br (35.8 vs. 5.7) has a higher S N 2 reaction rate than FCH 2 CH 2 Br.
) Consider Table 4 and suggest why FCH 2 CH 2 Br (5.7 vs. 1) has a higher S N 2 reaction rate than EtBr. 4b) Consider Table 4 and suggest why BrCH 2 CH 2 Br (35.8 vs. 5.7) has a higher S N 2 reaction rate than FCH 2 CH 2 Br.
) Consider Table 4 and suggest why FCH 2 CH 2 Br (5.7 vs. 1) has a higher S N 2 reaction rate than EtBr. 4b) Consider Table 4 and suggest why BrCH 2 CH 2 Br (35.8 vs. 5.7) has a higher S N 2 reaction rate than FCH 2 CH 2 Br.
In this study, the researcher compared S N 2 and E2 reaction rates for four substrates. Three of the substrates had a second halogen on the b position in the molecule. This work also compared the behavior of two nucleophiles: dianion I and II. You should read the abstract and look at Scheme 1 (p. 3082) and Table 4 (p. 3086).
Abstract: The gas-phase reactions of benzoate and phenolate containing dianions with a series of ‚-substituted alkyl bromides (X-CH2CH2Br, X ) H, F, Cl, Br) have been studied in a quadrupole ion trap mass spectrometer. Branching ratios between SN 2 and E2 products were measured and rate constants were determined. The ‚-halogens increase both the S N 2 and E2 rates, but the effect is greater for the latter process and therefore these substituents lead to an increase in the amount of elimination. The kinetic data for the SN 2 reactions can be analyzed via a two-parameter, linear free-energy relationship and the results indicate that field-effects (i.e., electron-withdrawing groups) strongly favor the reaction (FF ) 1.83). In contrast, analysis of the available condensed phase data for these substrates indicates that halogens strongly retard the reaction (FF ) -2.04). The dramatic reversal in substituent effects can be explained by a simple electrostatic model which suggests that solvation causes the system to shift to a more highly ionized SN2 transition state.
4a) Consider Table 4 and suggest why FCH 2 CH 2 Br (5.7 vs. 1) has a higher S N 2 reaction rate than EtBr.
4b) Consider Table 4 and suggest why BrCH 2 CH 2 Br (35.8 vs. 5.7) has a higher S N 2 reaction rate than FCH 2 CH 2 Br.
Transcribed Image Text:Scheme 1
c=c-O-co₂"
+ CH₂CH₂Br
SN2
=c-Q-caCH,CH, (3)
+ Br
ⒸC=CO-CO₂H
+ CH₂=CH₂ + Br
(4)
reactions of two dianion nucleophiles with a series of simple
halides and representative reactions are outlined in Scheme 1.
It can be seen that by starting with a doubly charged nucleophile,
each pathway leads to two ionic products. The alkylated (SN2)
and protonated (E2) nucleophiles still retain charges and
therefore can be used to identify the reaction mechanism (eqs
3 and 4).
A key assumption of the method is that the second charge
does not radically distort the reactivity of the nucleophilic center
so that the dianion can provide a good model for the properties
of a singly charged nucleophile. The potential problem with a
dianion is that it contains a significant amount of internal
electrostatic repulsion that will be released in the course of a
reaction with an alkyl halide as the charged species separate.
This issue has been explored by computational methods, and
for dianions with fairly large initial charge separations (~15 Å
or more), the second charge has only a modest effect on the
potential energy surface of a nucleophilic reaction such as an
SN2 substitution.22 The effect of the second charge is limited
because in reaching the transition state of an SN2 reaction, there
is a relatively small increase in the charge separation so only a
small amount of the internal electrostatic repulsion is released.
The majority of the internal electrostatic repulsion is released
after the transition state and has no effect on the kinetics or
product distribution of the reaction. As a result, dianion
nucleophiles can provide realistic models of singly charged
analogues in these reactions.23
In a previous communication, we demonstrated the utility
of the method and applied it to a series of alkyl bromides with
varying substitution patterns at the a-carbon (i.e., 1°, 2°, and
3º). Here, we describe an investigation of the effect of placing
electron-withdrawing groups at the ß-carbon of the substrate.
Specifically, the reactions of dianions I and II with a series of
O-co₂
2-haloethyl bromides (fluoro, chloro, and bromo) have been
examined. Both of these dianions were used in our previous
study and the diphenylacetylene framework provides a reason-
able charge separation (~14 Å) that limits the effects of internal
electrostatic repulsion. It is well-known that electron-withdraw-
ing groups at the B-carbon should enhance E2 rates because
they stabilize the incipient negative charge that develops on the
ß-carbon in the E2 transition state.2.24 Much of the previous
condensed phase work has focused on systems with either a
substituted aryl group or a powerful electron-withdrawing group
such as nitro or cyano at the ß-carbon.25-29 Less has been
reported on the effects of simple electron-withdrawing groups
such as halogens. In a very early study, Olivier and Weber
showed that 1,2-dibromoethane was over 100 times more
reactive than 1,1-dibromoethane under elimination conditions.30
Goering and Espy³1 also found that halogens and other groups
Energy
Rectants
Reactant
Complex
Transition
State
(8)
Products
(5)
v
Product
Complex
Reaction Coordinate
Figure 1. Double-well potential for gas-phase ion-molecule reac-
tions: (a) positive activation energy (solid line) and (b) negative
activation energy (dashed line).
at the B-carbon increase the E2 rate. In contrast, Okamoto and
co-workers³2 found that alkoxy and chloro substituents at the
ß-carbon caused a reduction in E2 rates in aqueous solution.
They rationalized this result as a steric effect. As for SN2
reactions, there is evidence from condensed phase studies that
electron-withdrawing groups at the ß-carbon (including halo-
gens) lead to rate reductions. 32-35 In the present study, we test
the validity of the generalization that electron-withdrawing
groups at the ß-carbon increase E2 rates and decrease SN2 rates
by determining the SN2 and E2 rate constants of the reactions
of the 2-haloethyl bromides with I and II. In addition, we
evaluate the relative abilities of the various halogens to stabilize/
destabilize the transition states.
To support the experimental work, ab initio calculations have
been completed. Given the size of the dianions, it is impractical
to pursue calculations on the actual experimental systems.
Instead, we have used two types of model systems. The reaction
of acetate with the 2-haloethyl bromides provides a reasonable
model of the reactions of the benzoate dianion, I. The other
model system uses methoxide as the nucleophile. Of course,
methoxide does not provide a realistic model of the phenolate
nucleophile, II, because it is a much stronger gas-phase base,
but it does offer insight into the effects of base strength and
charge localization on the competition between SN2 and E2
reactions in the B-substituted systems. The SN2 and E2 reactions
involve a single barrier and lead to a double-well potential
energy surface originally proposed by Brauman (Figure 1). For
each reaction, we have computationally characterized four
stationary points on the surface: the reactants, the reactant ion-
dipole complex, the transition state, and the products. On this
type of gas-phase potential energy surface, the activation energy
is defined as the energy difference between the transition state
and the separated reactants. As a result, positive (Figure la)
and negative (Figure 1b) activation energies are possible.
Experimental Section
Mass Spectrometry. All experiments were completed in a modified
Finnigan LCQ quadrupole ion trap mass spectrometer equipped with
(25) Bunnett, J. F.; Sridharan, S.; Cavin, W. P. J. Org. Chem. 1979, 44,
1463.
(26) Crosby, J.; Stirling, C. J. M. J. Am. Chem. Soc. 1968, 90, 6869.
(27) Gandler, J. R.; Yokoyama, T. J. Am. Chem. Soc. 1984, 106, 130.
(28) Marshall, D. R.; Thomas, P. J.; Stirling, C. J. M. J. Chem. Soc.,
Perkin Trans. 2 1977, 1914.
(29) Saunders: W. H., Jr. Acc. Chem. Res. 1976, 9, 19.
(30) Olivier, S. C. J.; Weber, A. P. Recl. Trav. Chim. 1934, 53, 1087.
(31) Goering, H. L.; Espy, H. H. J. Am. Chem. Soc. 1956, 78, 1454.
(32) Okamoto, K.; Kita, T.; Araki, K.; Shingu, H. Bull. Chem. Soc. Jpn.
1967, 40, 1913.
Transcribed Image Text:Table 4. Relative Rates for the SN2 and E2 Reactions of Dianions
with Alkyl Bromides
I
CH3CH₂Br
FCH₂CH₂Br
E2
SN2
1.7
9.3
0.0
0.0
3.5
17.1
0.4
SN2
1.0
5.7
CICH₂CH₂Br
13.7
BrCH₂CH₂Br
35.8
EtCH₂CH₂Br
2.8
"The yield was too low for accurate partial rate measurements.
II
a
a
E2
0.1
4.7
530
780
Expert Solution
This question has been solved!
Explore an expertly crafted, step-by-step solution for a thorough understanding of key concepts.
Need a deep-dive on the concept behind this application? Look no further. Learn more about this topic, chemistry and related others by exploring similar questions and additional content below.